






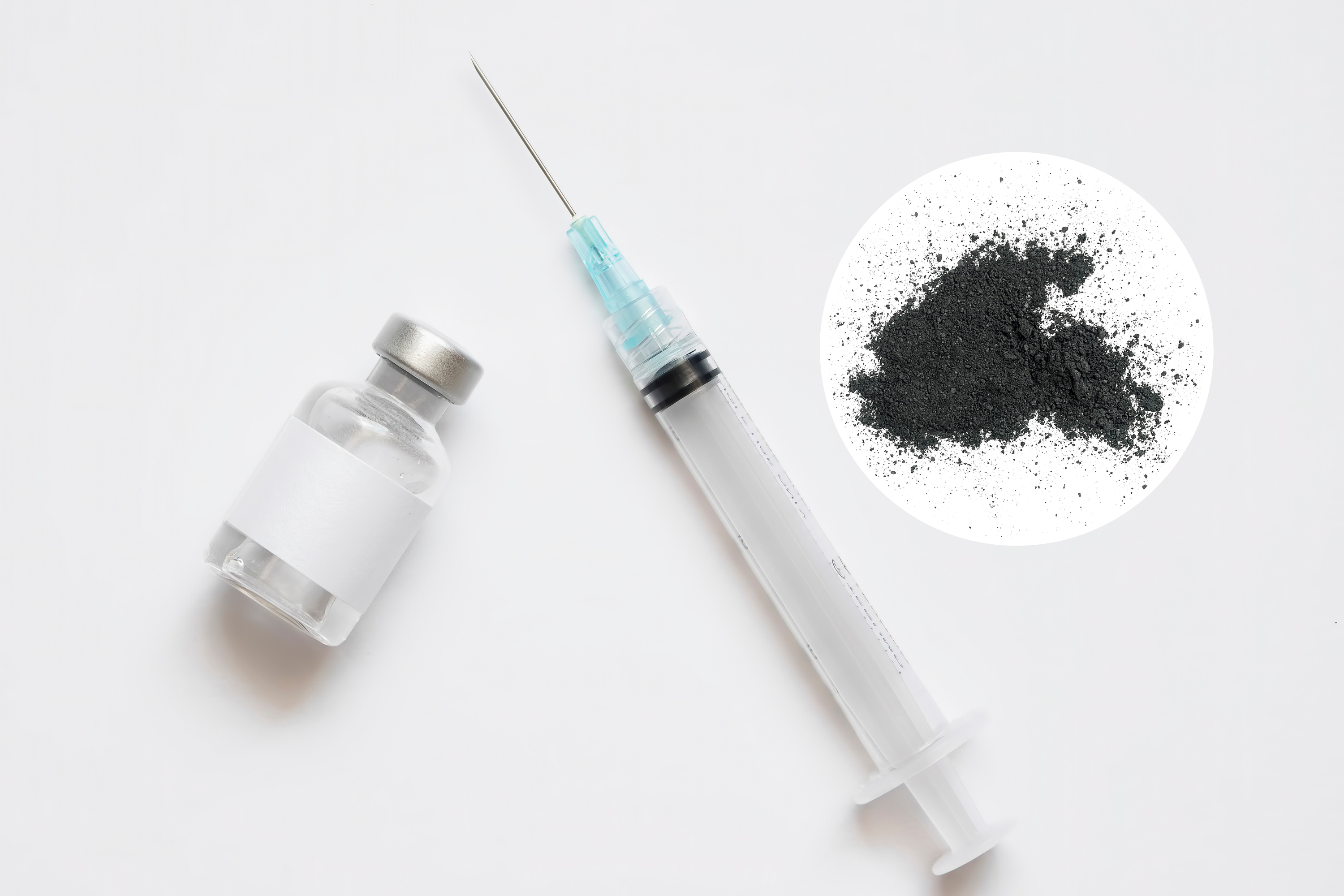
A nationwide voluntary recall of phenylephrine hydrochloride injection, a drug used to combat low blood pressure, has been initiated due to the discovery of black particulate matter in a single vial of the product. This contamination poses a significant health risk, as injecting particulate matter can lead to localized reactions like swelling and irritation, or more severe complications such as stroke and death if the particles block blood vessels in vital organs like the heart, lungs, or brain. The recall, announced by both the manufacturer, Provepharm, Inc., and the U.S. Food and Drug Administration (FDA), underscores the potential danger of administering contaminated medications and highlights the importance of vigilant quality control measures within the pharmaceutical industry. While no adverse events related to the contaminated lot have been reported, the recall aims to prevent potential harm by removing the affected product from circulation.
The recalled product, specifically the 10mg/ml concentration of phenylephrine hydrochloride injection, is identified by lot code “24020027,” an expiry date of December 2025, and NDC “81284-213-01.” This specific lot was distributed to wholesalers, distributors, compounding companies, and hospitals, emphasizing the widespread potential reach of the contaminated vials. Provepharm has partnered with Sedgwick, a recall provider, to manage the logistics of notifying affected parties, coordinating returns, and ensuring the safe disposal of the recalled product. This collaborative approach demonstrates a commitment to containing the issue and minimizing potential harm to patients.
The discovery of the particulate matter stemmed from a customer complaint filed by a pharmacy, highlighting the crucial role of post-market surveillance in identifying potential drug safety issues. This proactive reporting mechanism allowed for a swift response, preventing further distribution of the contaminated lot and minimizing potential exposure. The FDA’s subsequent announcement reinforced the seriousness of the issue, providing detailed information about the recall and urging healthcare professionals and patients to take appropriate action. This collaborative approach between manufacturers, regulators, and healthcare providers is vital in ensuring patient safety.
The FDA advises anyone possessing the recalled phenylephrine hydrochloride injection to immediately discontinue its use and return the product to Sedgwick. Clear instructions and contact information have been provided to facilitate the return process, including a dedicated phone line and email address managed by Sedgwick. This streamlined process aims to simplify the return procedure and ensure efficient removal of the contaminated product from the market. Furthermore, Provepharm has established a dedicated contact point for individuals with medical questions related to the recalled product, demonstrating a commitment to patient care and providing a channel for addressing specific concerns.
The potential consequences of administering contaminated injections underscore the critical importance of rigorous quality control measures throughout the pharmaceutical supply chain. From manufacturing to distribution, stringent protocols must be in place to prevent contamination and ensure the integrity of injectable medications. This incident serves as a reminder of the potential for even minute contaminants to pose serious health risks and reinforces the need for continuous monitoring and improvement of manufacturing processes. The collaborative response between Provepharm, the FDA, and Sedgwick exemplifies the importance of a multi-pronged approach to product recalls, ensuring effective communication and efficient removal of potentially harmful products.
This recall also highlights the importance of patient vigilance and reporting suspected issues with medications. The initial discovery of the contamination stemmed from a customer complaint, demonstrating the valuable role patients and healthcare professionals play in post-market surveillance. The FDA encourages anyone experiencing adverse reactions or quality problems with any medication to report it through their MedWatch Adverse Event Reporting program. This reporting system serves as a crucial tool for identifying potential safety issues and ensuring timely interventions to protect public health. By working together, patients, healthcare providers, and regulatory agencies can contribute to a safer medication environment.






